This is the abstract class 'omics', contains a variety of methods that are inherited and applied in the omics classes: metagenomics and proteomics.
Details
Every class is created with the R6Class method. Methods are either public or private, and only the public components are inherited by other omic classes. The omics class by default uses a sparseMatrix and data.table data structures for quick and efficient data manipulation and returns the object by reference, same as the R6 class. The method by reference is very efficient when dealing with big data.
References
Aitchison, J. (1986) The Statistical Analysis of Compositional Data. Chapman and Hall, London, 416 p.
Active bindings
metaDataA data.table with
SAMPLE_IDcolumn.featureDataA data.table with
FEATURE_IDcolumn.countDataA dense or sparse Matrix.
Methods
omics$new()
Wrapper function that is inherited and adapted for each omics class.
The omics classes requires a metadata samplesheet, that is validated by the metadata_schema.json.
It requires a column SAMPLE_ID and optionally a SAMPLEPAIR_ID can be supplied.
The SAMPLE_ID will be used to link the metaData to the countData, and will act as the key during subsetting of other columns.
To create a new object use new() method. Do notice that the abstract class only checks if the metadata is valid!
The countData and featureData will not be checked, these are handled by the sub-classes.
Using the omics class to load your data is not supported and still experimental.
Usage
omics$new(countData = NULL, featureData = NULL, metaData = NULL)Arguments
countDataA path to an existing file or a dense/sparse Matrix format.
featureDataA path to an existing file, data.table or data.frame.
metaDataA path to an existing file, data.table or data.frame.
omics$copy()
Create a copy of the object-class
This method is very similar to the existing clone() function, except it also resets the back-up of the OmicFlow data types that is invoked with reset()
Examples
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
obj <- omics$new(
metaData = metadata_file,
countData = counts_file
)
# Perform a modification and copy
obj$scale()
cloned <- obj$copy(deep=TRUE)
cloned$scale(method = "clr")
cloned$reset() # resets to data after clone creation.omics$validate()
Validates an input metadata against the JSON schema. The metadata should look as follows and should not contain any empty spaces.
For example; 'sample 1' is not allowed, whereas 'sample1' is allowed!
Acceptable column headers:
SAMPLE_ID (required)
SAMPLEPAIR_ID (optional)
CONTRAST_ (optional), used for
autoFlow().VARIABLE_ (optional), not supported yet.
This function is used during the creation of a new object via new() to validate the supplied metadata
via a filepath or existing data.table or data.frame.
omics$print()
Displays parameters of the omics class via stdout.
Examples
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
obj <- omics$new(
metaData = metadata_file,
countData = counts_file
)
# method 1 to call print function
obj
# method 2 to call print function
obj$print()omics$reset()
Upon creation of a new omics object a small backup of the original data is created.
Since modification of the object is done by reference and duplicates are not made, it is possible to reset changes to the class.
The methods from the abstract class omics also contains a private method to prevent any changes to the original object when using methods such as ordination alpha_diversity or foldchange.
Examples
library(ggplot2)
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
taxa <- omics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file
)
# Performs modifications
taxa$scale(transform = log2)
# resets
taxa$reset()
# An inbuilt reset function prevents unwanted modification to the taxa object.
taxa$rankstat(feature_ranks = c("Kingdom", "Phylum", "Family", "Genus", "Species"))omics$removeNAs()
Remove NAs from metaData and updates the countData.
Arguments
columnThe column from where NAs should be removed, this can be either a wholenumbers or characters. Vectors are also supported.
Examples
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
obj$removeNAs(column = "treatment")omics$feature_subset()
Feature subset (based on featureData), automatically applies data synchronization.
Arguments
...Expressions that return a logical value, and are defined in terms of the variables in
featureData. Only rows for which all conditions evaluate to TRUE are kept.
Examples
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
obj$feature_subset(Genus == "Pseudomonas")omics$sample_subset()
Sample subset (based on metaData), automatically applies synchronization.
Arguments
...Expressions that return a logical value, and are defined in terms of the variables in
metaData. Only rows for which all conditions evaluate to TRUE are kept.
Examples
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
obj$sample_subset(treatment == "tumor")omics$samplepair_subset()
Samplepair subset (based on metaData), automatically applies synchronization.
omics$feature_merge()
Agglomerates features by column, automatically applies synchronization.
Arguments
feature_rankA character value or vector of columns to aggregate from the
featureData.feature_filterA character value or vector of characters to remove features via regex pattern.
Examples
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
obj$feature_merge(feature_rank = c("Kingdom", "Phylum"))
obj$feature_merge(feature_rank = "Genus", feature_filter = c("uncultured", "metagenome"))omics$scale()
Feature scaling on the countData. The scale function is able to apply transformations element-wise on the positive values, (optional: add pseudocounts) and perform normalisation or standardisation methods.
Usage
omics$scale(
method = "tss",
transform = NULL,
base = exp(1),
pseudocount = NULL
)Arguments
methodA character to choose a standardisation/normalisation method, options:
tss,clr,binary,hellinger,none(default:"tss").transformA function to apply on the positive values of
countData, skip standardisation/normalisation withmethod = "none"(default:NULL).baseInput for log to use natural logarithmic scale, log2, log10 or other (default:
exp(1)) in CLR.pseudocountA numeric value to replace zero's (default:
NULL).
Examples
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
# standard relative abundance computation
obj$scale()
# CLR
obj$reset()
obj$scale(method = "clr")
# transform
obj$reset()
obj$scale(method = "none", transform = log2)omics$rankstat()
Rank statistics based on featureData
Arguments
feature_ranksA vector of characters or integers that match the
featureData.uniqueA boolean value to display only unique entries in
feature_ranks.
Details
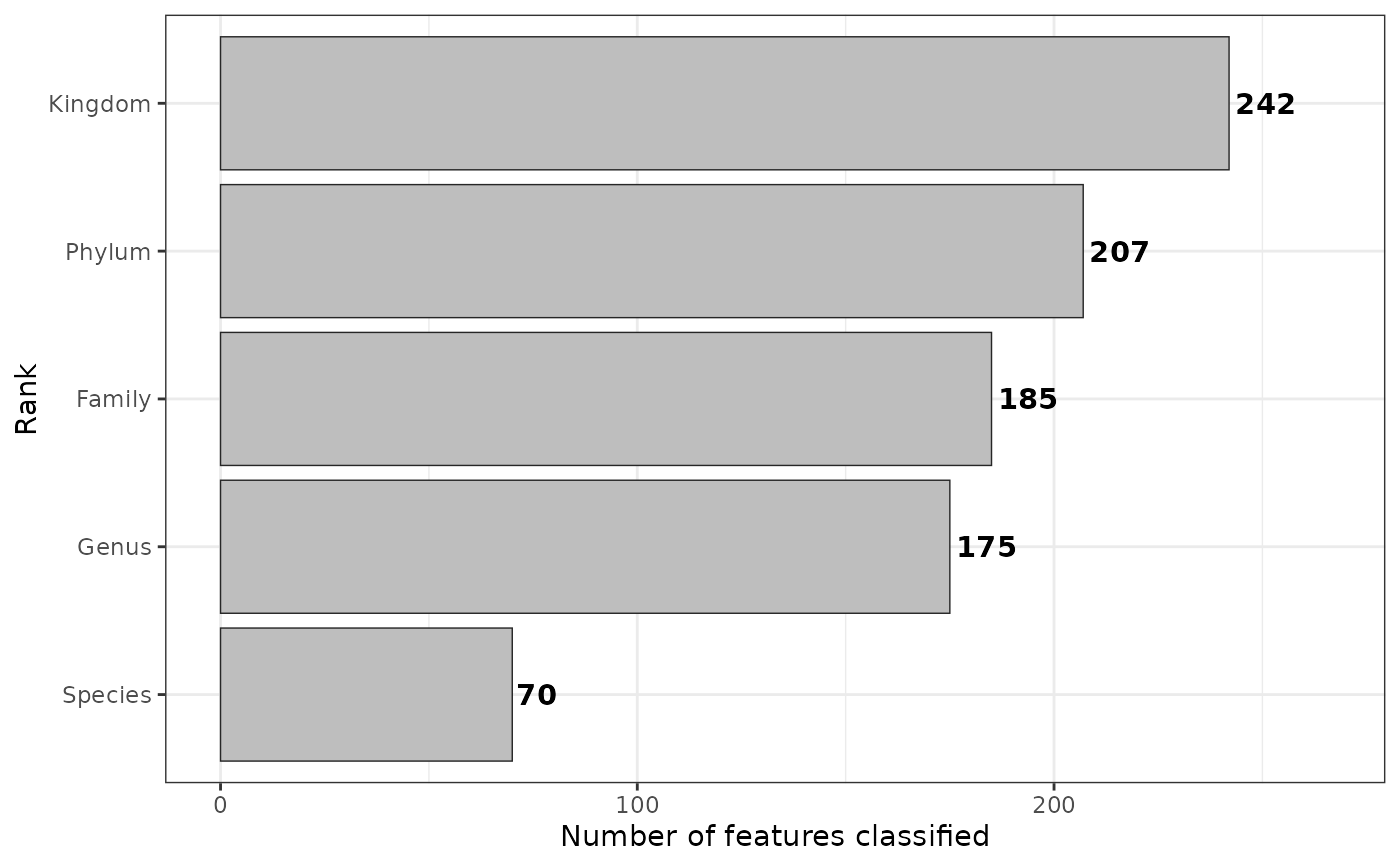
Counts the number of features identified for each column, for example in case of 16S metagenomics it would be the number of OTUs or ASVs on different taxonomy levels.
Returns
A ggplot object.
Examples
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
plt <- obj$rankstat(feature_ranks = c("Kingdom", "Phylum", "Family", "Genus", "Species"))
pltomics$alpha_diversity()
Alpha diversity based on diversity
Usage
omics$alpha_diversity(
col_name,
metric = c("shannon", "invsimpson", "simpson"),
Brewer.palID = "Set2",
group_by = NULL,
evenness = FALSE,
paired = FALSE,
p.adjust.method = "fdr"
)Arguments
col_nameA character variable from the
metaData.metricAn alpha diversity metric as input to diversity.
Brewer.palIDA character name for the palette set to be applied, see brewer.pal or colormap.
group_byA column name to perform grouped statistical test in diversity_plot (default: NULL).
evennessA boolean wether to divide diversity by number of species, see specnumber.
pairedA boolean value to perform paired analysis in wilcox.test and samplepair subsetting via
samplepair_subset()p.adjust.methodA character variable to specify the p.adjust.method to be used, default is 'fdr'.
Returns
A list of components:
divA data.frame from diversity.statsA pairwise statistics from pairwise_wilcox_test.plotA ggplot object.
Examples
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
plt <- obj$alpha_diversity(col_name = "treatment",
metric = "shannon")omics$composition()
Creates a table most abundant compositional features. Also assigns a color blind friendly palette for visualizations.
Usage
omics$composition(
feature_rank,
feature_filter = NULL,
col_name = NULL,
feature_top = c(10, 15),
Brewer.palID = "RdYlBu"
)Arguments
feature_rankA character variable in
featureDatato aggregate viafeature_merge().feature_filterA character or vector of characters to removes features by regex pattern.
col_nameOptional, a character or vector of characters to add to the final compositional data output.
feature_topA wholenumber of the top features to visualize, the max is 15, due to a limit of palettes.
Brewer.palIDA character name for the palette set to be applied, see brewer.pal or colormap.
Examples
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
result <- obj$composition(feature_rank = "Genus",
feature_filter = c("uncultured"),
feature_top = 10)
plt <- composition_plot(data = result$data,
palette = result$palette,
feature_rank = "Genus")omics$distance()
Compute a distance metric from countData
Usage
omics$distance(
metric,
weighted = TRUE,
threads = 1,
normalized = TRUE,
base = exp(1)
)Arguments
metricA dissimilarity metric to be applied on the
countData, thus far supports 'bray', 'jaccard', 'cosine', 'manhattan', 'aitchison', 'euclidean', 'jsd' (jensen-shannon divergence), 'canberra' and 'unifrac' when a tree is provided viatreeData, seedistance().weightedA boolean value, to use abundances (
weighted = TRUE) or absence/presence (weighted=FALSE) (default: TRUE).threadsA wholenumber, indicating the number of threads to use (Default: 1).
normalizedA boolean value, whether to normalize weighted UniFrac distances to be between 0 and 1. Unweighted UniFrac is always normalized (default: TRUE).
baseInput for log to use natural logarithmic scale, log2, log10 or other (default:
exp(1)).pseudocountA numeric value to replace zero's, used in
scale()(default:1e-15).
Returns
A column x column dist object.
Examples
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file
)
obj$feature_subset(Kingdom == "Bacteria")
dist <- obj$distance(metric = "bray")omics$ordination()
Ordination of countData with statistical testing.
Usage
omics$ordination(
metric = "bray",
method = c("pcoa", "nmds"),
group_by,
distmat = NULL,
weighted = TRUE,
threads = 1,
perm_design = NULL,
perm = 999
)Arguments
metricA dissimilarity or similarity metric to be applied on the
countData, thus far supports 'bray', 'jaccard', 'cosine', 'manhattan', 'jsd' (jensen-shannon divergence), 'canberra' and 'unifrac' when a tree is provided viatreeData, seedistance().methodOrdination method, supports "pcoa" and "nmds", see wcmdscale.
group_byA character variable in
metaDatato be used for the pairwise_adonis or pairwise_anosim statistical test.distmatweightedA boolean value, whether to compute weighted or unweighted dissimilarities (default:
TRUE).threadsA wholenumber, indicating the number of threads to use (Default: 1).
perm_designA function that takes
metaDataand constructs a permutation design with how (default:NULL).permA wholenumber, number of permutations to compare against the null hypothesis of adonis2 and anosim (default:
perm=999).
Returns
A list of components:
distmatA distance dissimilarity in matrix format.statsA statistical test as a data.frame.pcsprincipal components as a data.frame.scree_plotA ggplot object.anova_plotA ggplot object.scores_plotA ggplot object.
Examples
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
pcoa_plots <- obj$ordination(metric = "bray",
method = "pcoa",
group_by = "treatment",
weighted = TRUE)
pcoa_plotsomics$foldchange()
Differential feature expression (DFE) on log-transformed values for both paired and non-paired test.
The function performs feature agglomeration, subsetting to remove NAs in condition.group and finding samplepairs. It expects that the data is already log-transformed, this can be accomplished via scale()
Usage
omics$foldchange(
condition.group,
condition_A,
condition_B,
group_by = NULL,
feature_rank = "FEATURE_ID",
feature_filter = NULL,
paired = FALSE,
pvalue.threshold = 0.05,
logfold.threshold = 0.06,
abundance.threshold = 0
)Arguments
condition.groupA character variable of an existing column name in
metaData, wherein the conditions A and B are located.condition_AA character value or vector of characters.
condition_BA character value or vector of characters.
group_byA character variable of an existing column in
metaDatato split the table in chunks prior to fold-change computation (default:NULL). When disabled then column names will end with_in_all.feature_rankA character or vector of characters in the
featureDatato aggregate viafeature_merge()(default:"FEATURE_ID").feature_filterA character or vector of characters to remove features via regex pattern (default:
NULL).pairedA boolean value, the paired is only applicable when a
SAMPLEPAIR_IDcolumn exists within themetaData. See wilcox.test andsamplepair_subset().pvalue.thresholdA numeric value used as a p-value threshold to label and color significant features (default: 0.05).
logfold.thresholdA numeric value used as a fold-change threshold to label and color significantly expressed features (default: 0.06).
abundance.thresholdA numeric value used as an abundance threshold to size the scatter dots based on their mean abundance (default: 0.01).
Returns
dataA long data.table table.volcano_plotA ggplot object.AA data.table table for (each) condition ABA data.table table for (each) condition B
Examples
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- omics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file
)
obj$scale(method = "clr")
dfe <- obj$foldchange(feature_rank = "Genus",
paired = FALSE,
condition.group = "treatment",
condition_A = c("healthy"),
condition_B = c("tumor"))omics$autoFlow()
Automated Omics Analysis based on the metaData, see validate().
For now only works with headers that start with prefix CONTRAST_. If the data is from the class omics or proteomics than FDR adjusted p-values are computed for the volcano plots. Log-transformed values will lead to the skipping of composition() and alpha_diversity() methods.
Usage
omics$autoFlow(
feature_contrast = "FEATURE_ID",
feature_filter = NULL,
feature_ranks = NULL,
distance_metrics = c("bray"),
distmat = NULL,
weighted = TRUE,
pvalue.threshold = 0.05,
logfold.threshold = 1,
abundance.threshold = 0.01,
perm = 999,
threads = 1,
report = TRUE,
filename = paste0(getwd(), "/report.html")
)Arguments
feature_contrastA character vector of feature columns in the
featureDatato aggregate viafeature_merge()(default:"FEATURE_ID").feature_filterA character vector to filter unwanted features, (default:
NULL).feature_ranksA character vector as input to
rankstat()(default:NULL).distance_metricsA character vector specifying what (dis)similarity metrics to use (default:
c("bray")) When you are working with log-transformed data it is advised to use theeuclidean.distmatA path to an existing file or a dense/sparse Matrix format (default:
NULL).weightedA boolean value, whether to compute weighted or unweighted dissimilarities (default:
TRUE).pvalue.thresholdA numeric value, the p-value is used to include/exclude composition and foldchanges plots coming from alpha- and beta diversity analysis (default: 0.05).
logfold.thresholdA numeric value used as a fold-change threshold to label and color significantly expressed features, see
foldchange()(Default: 1).abundance.thresholdA numeric value used as an abundance threshold to size the scatter dots based on their mean abundance, see
foldchange()(default: 0.01).permA wholenumber, number of permutations to compare against the null hypothesis of adonis2 or anosim (default: 999).
threadsNumber of threads to use, only used in
distance()when distmat is not supplied (default: 1).reportA boolean value to create a HTML markdown report (default:
FALSE). IfFALSEa nested list of the plots and data is returned.filenameA character to name the HTML report to be saved in the current working directory (default:
paste0(getwd(), "/report.html")). Thegetwd()is required for rmarkdown to save it in the right path.
Examples
## ------------------------------------------------
## Method `omics$copy()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
obj <- omics$new(
metaData = metadata_file,
countData = counts_file
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ countData is loaded.
#> ! Created placeholder featureData.
# Perform a modification and copy
obj$scale()
cloned <- obj$copy(deep=TRUE)
cloned$scale(method = "clr")
cloned$reset() # resets to data after clone creation.
## ------------------------------------------------
## Method `omics$print()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
obj <- omics$new(
metaData = metadata_file,
countData = counts_file
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ countData is loaded.
#> ! Created placeholder featureData.
# method 1 to call print function
obj
#>
#> ── <omics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 0 attributes × 242 features
# method 2 to call print function
obj$print()
#>
#> ── <omics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 0 attributes × 242 features
## ------------------------------------------------
## Method `omics$reset()`
## ------------------------------------------------
library(ggplot2)
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
taxa <- omics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
# Performs modifications
taxa$scale(transform = log2)
# resets
taxa$reset()
# An inbuilt reset function prevents unwanted modification to the taxa object.
taxa$rankstat(feature_ranks = c("Kingdom", "Phylum", "Family", "Genus", "Species"))
 ## ------------------------------------------------
## Method `omics$removeNAs()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
obj$removeNAs(column = "treatment")
## ------------------------------------------------
## Method `omics$feature_subset()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
obj$feature_subset(Genus == "Pseudomonas")
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 3 samples
#> countData: 3 samples × 4 features
#> featureData: 7 attributes × 4 features
## ------------------------------------------------
## Method `omics$sample_subset()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
obj$sample_subset(treatment == "tumor")
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 2 samples
#> countData: 2 samples × 115 features
#> featureData: 7 attributes × 115 features
## ------------------------------------------------
## Method `omics$feature_merge()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
obj$feature_merge(feature_rank = c("Kingdom", "Phylum"))
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 28 features
#> featureData: 7 attributes × 28 features
obj$feature_merge(feature_rank = "Genus", feature_filter = c("uncultured", "metagenome"))
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 20 features
#> featureData: 7 attributes × 20 features
## ------------------------------------------------
## Method `omics$scale()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
# standard relative abundance computation
obj$scale()
# CLR
obj$reset()
obj$scale(method = "clr")
# transform
obj$reset()
obj$scale(method = "none", transform = log2)
## ------------------------------------------------
## Method `omics$rankstat()`
## ------------------------------------------------
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
plt <- obj$rankstat(feature_ranks = c("Kingdom", "Phylum", "Family", "Genus", "Species"))
plt
## ------------------------------------------------
## Method `omics$removeNAs()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
obj$removeNAs(column = "treatment")
## ------------------------------------------------
## Method `omics$feature_subset()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
obj$feature_subset(Genus == "Pseudomonas")
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 3 samples
#> countData: 3 samples × 4 features
#> featureData: 7 attributes × 4 features
## ------------------------------------------------
## Method `omics$sample_subset()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
obj$sample_subset(treatment == "tumor")
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 2 samples
#> countData: 2 samples × 115 features
#> featureData: 7 attributes × 115 features
## ------------------------------------------------
## Method `omics$feature_merge()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
obj$feature_merge(feature_rank = c("Kingdom", "Phylum"))
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 28 features
#> featureData: 7 attributes × 28 features
obj$feature_merge(feature_rank = "Genus", feature_filter = c("uncultured", "metagenome"))
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 20 features
#> featureData: 7 attributes × 20 features
## ------------------------------------------------
## Method `omics$scale()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
# standard relative abundance computation
obj$scale()
# CLR
obj$reset()
obj$scale(method = "clr")
# transform
obj$reset()
obj$scale(method = "none", transform = log2)
## ------------------------------------------------
## Method `omics$rankstat()`
## ------------------------------------------------
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
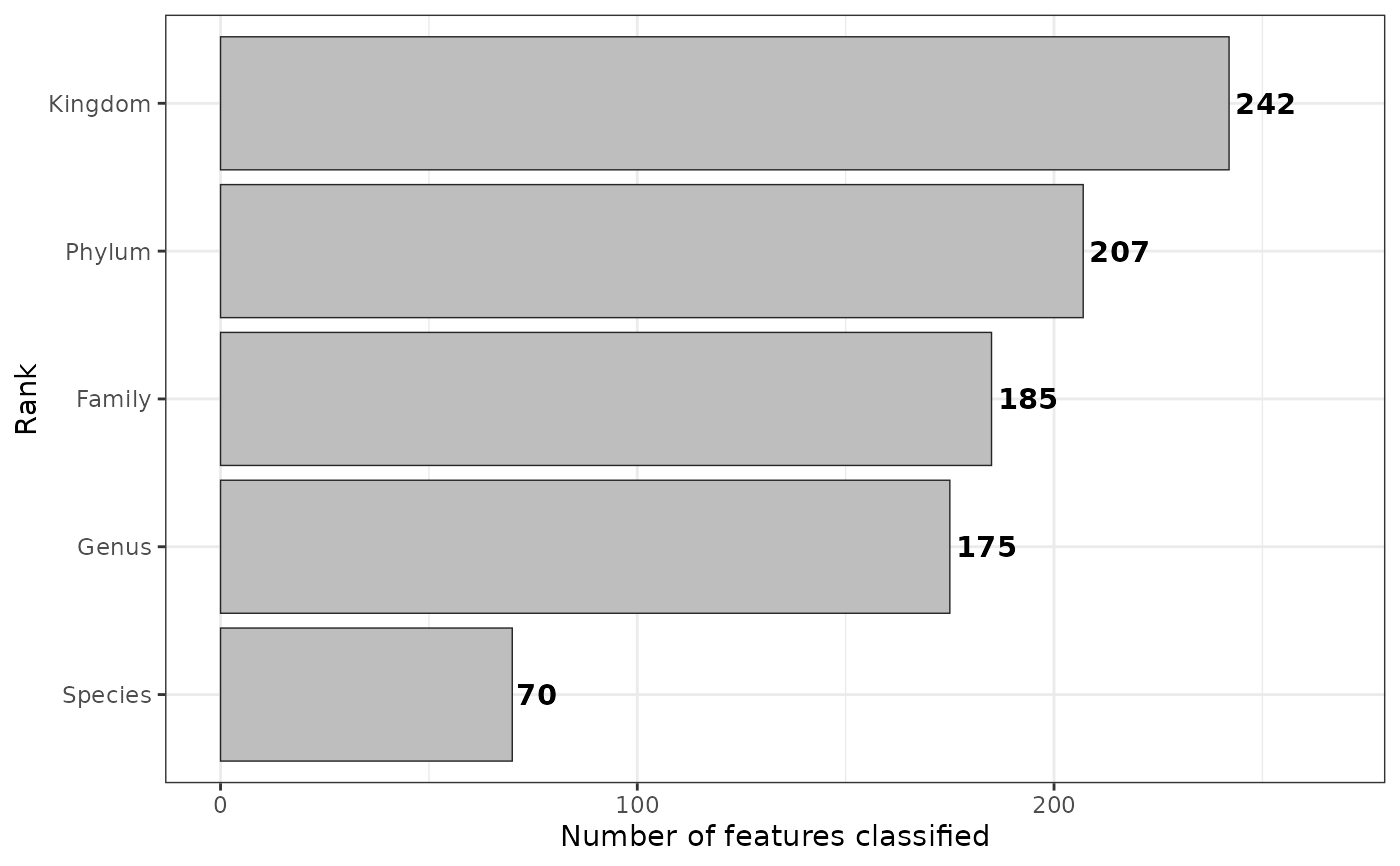
plt <- obj$rankstat(feature_ranks = c("Kingdom", "Phylum", "Family", "Genus", "Species"))
plt
 ## ------------------------------------------------
## Method `omics$alpha_diversity()`
## ------------------------------------------------
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
plt <- obj$alpha_diversity(col_name = "treatment",
metric = "shannon")
## ------------------------------------------------
## Method `omics$composition()`
## ------------------------------------------------
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
result <- obj$composition(feature_rank = "Genus",
feature_filter = c("uncultured"),
feature_top = 10)
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 63 features
#> featureData: 7 attributes × 63 features
plt <- composition_plot(data = result$data,
palette = result$palette,
feature_rank = "Genus")
## ------------------------------------------------
## Method `omics$distance()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
obj$feature_subset(Kingdom == "Bacteria")
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 185 features
#> featureData: 7 attributes × 185 features
dist <- obj$distance(metric = "bray")
## ------------------------------------------------
## Method `omics$ordination()`
## ------------------------------------------------
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
pcoa_plots <- obj$ordination(metric = "bray",
method = "pcoa",
group_by = "treatment",
weighted = TRUE)
#> 'nperm' >= set of all permutations: complete enumeration.
#> Set of permutations < 'minperm'. Generating entire set.
pcoa_plots
#> $dist
#> S100 S103 S115 S120
#> S100 0.0000000 1.0000000 0.8845188 1.0000000
#> S103 1.0000000 0.0000000 1.0000000 0.9470058
#> S115 0.8845188 1.0000000 0.0000000 1.0000000
#> S120 1.0000000 0.9470058 1.0000000 0.0000000
#>
#> $anova_data
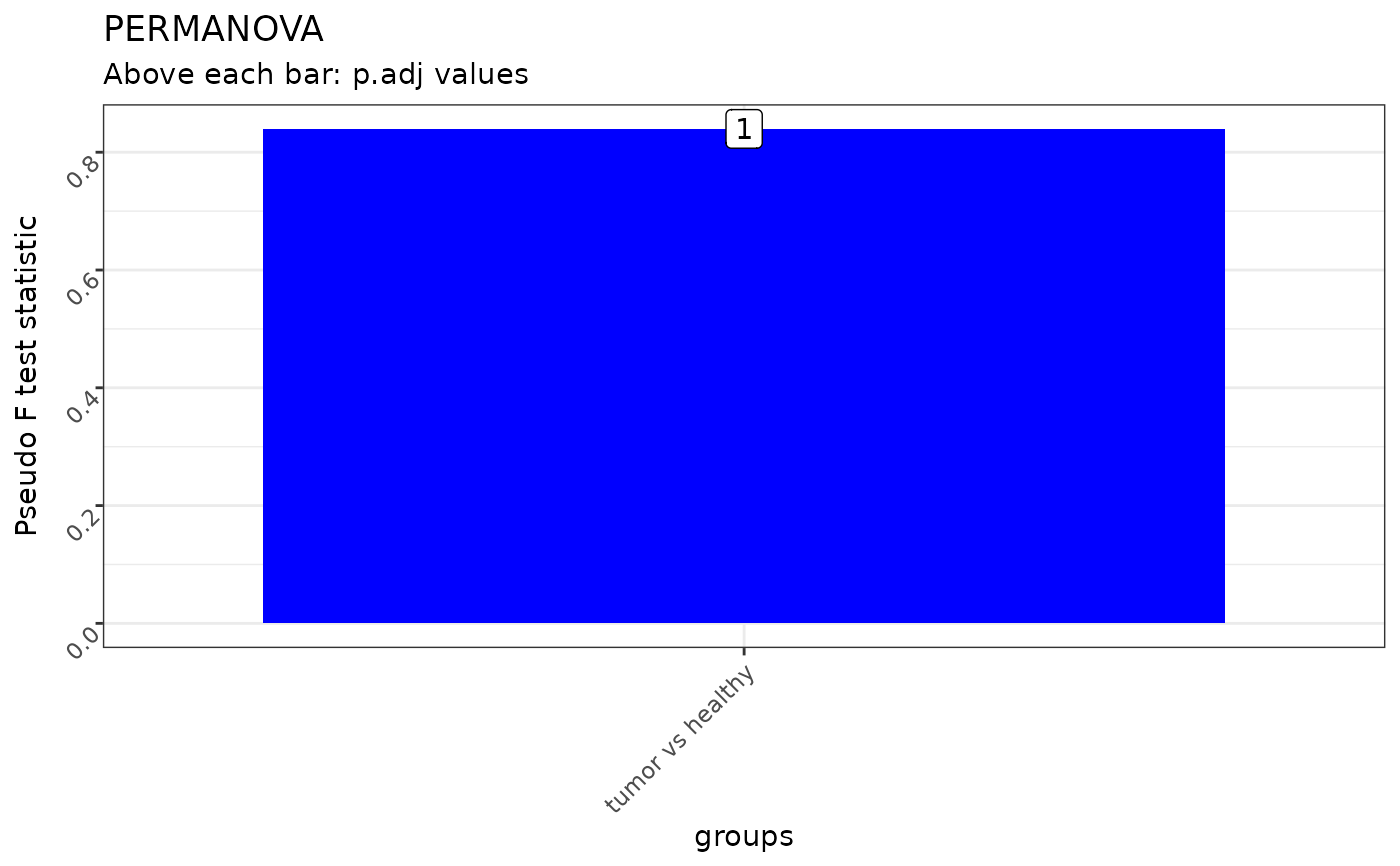
#> pairs Df SumsOfSqs F.Model R2 p.value p.adj
#> 1 tumor vs healthy 1 0.4197984 0.8395968 0.2956747 1 1
#>
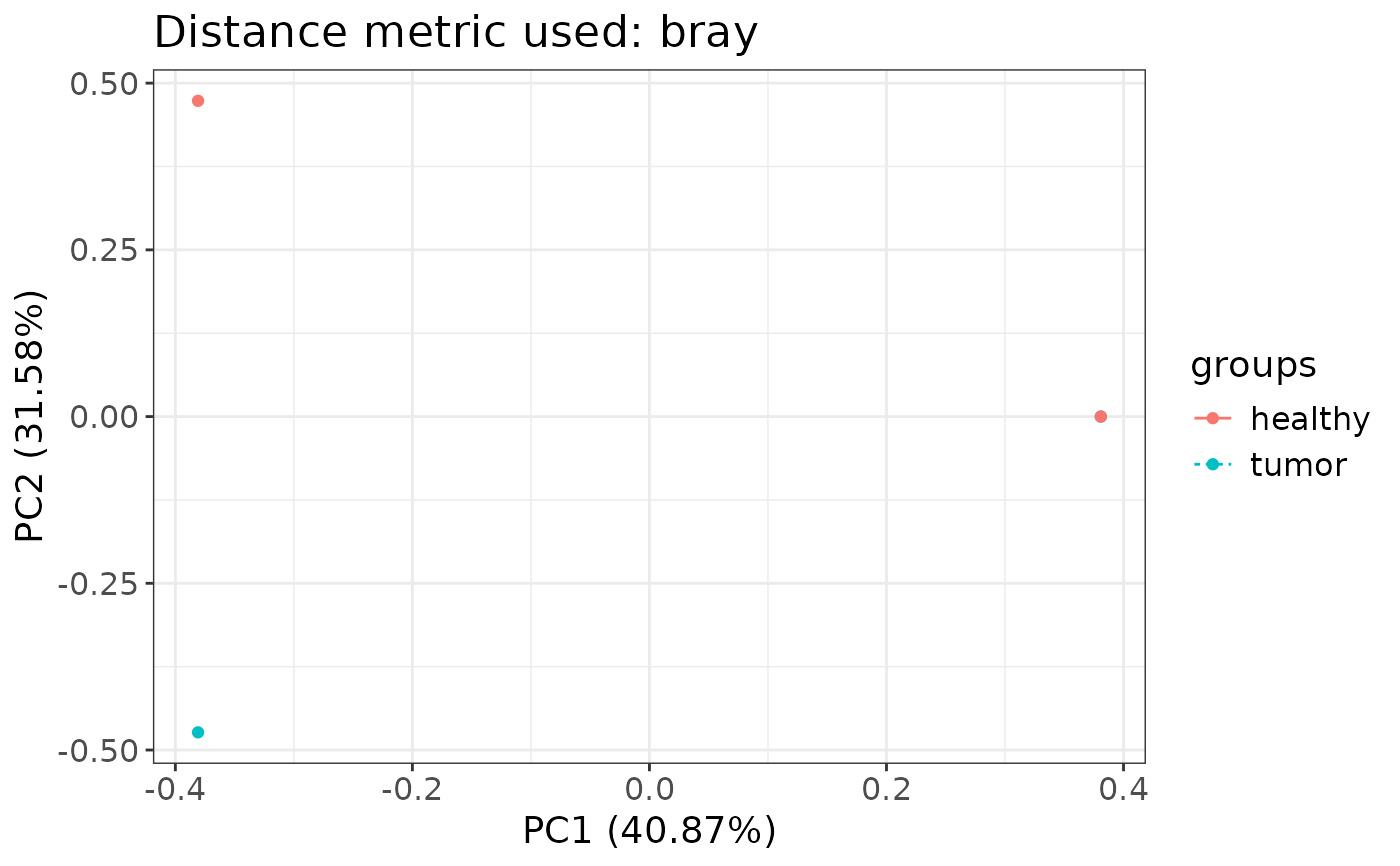
#> $pcs
#> PC1 PC2 PC3 groups samples
#> <num> <num> <num> <char> <char>
#> 1: 0.3808548 0.000000e+00 4.422594e-01 tumor 1
#> 2: -0.3808548 -4.735029e-01 -2.256762e-16 tumor 2
#> 3: 0.3808548 -2.787913e-17 -4.422594e-01 healthy 3
#> 4: -0.3808548 4.735029e-01 -2.603956e-16 healthy 4
#>
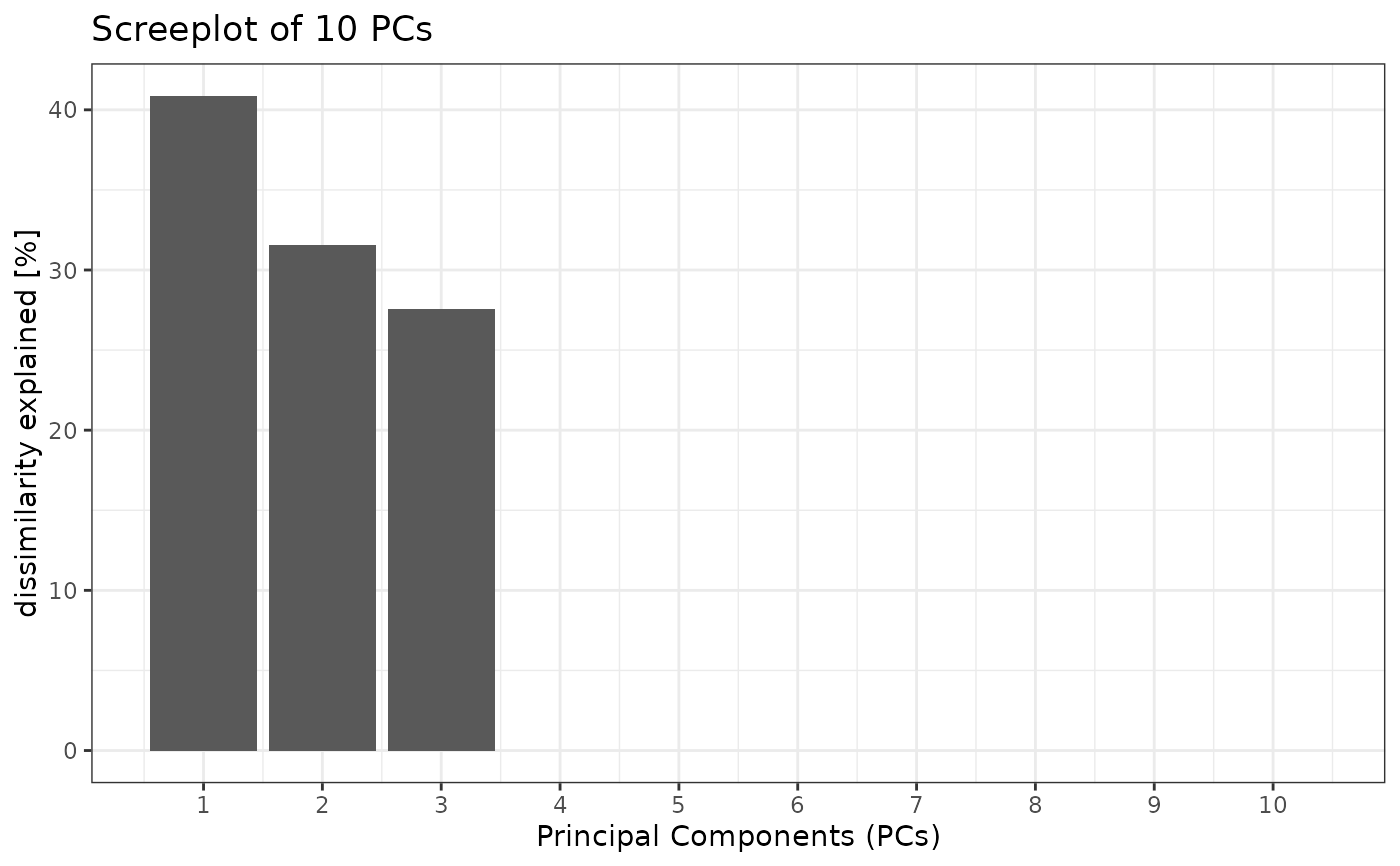
#> $scree_plot
#> Warning: Removed 7 rows containing missing values or values outside the scale range
#> (`geom_col()`).
## ------------------------------------------------
## Method `omics$alpha_diversity()`
## ------------------------------------------------
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
plt <- obj$alpha_diversity(col_name = "treatment",
metric = "shannon")
## ------------------------------------------------
## Method `omics$composition()`
## ------------------------------------------------
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
result <- obj$composition(feature_rank = "Genus",
feature_filter = c("uncultured"),
feature_top = 10)
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 63 features
#> featureData: 7 attributes × 63 features
plt <- composition_plot(data = result$data,
palette = result$palette,
feature_rank = "Genus")
## ------------------------------------------------
## Method `omics$distance()`
## ------------------------------------------------
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
obj$feature_subset(Kingdom == "Bacteria")
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 185 features
#> featureData: 7 attributes × 185 features
dist <- obj$distance(metric = "bray")
## ------------------------------------------------
## Method `omics$ordination()`
## ------------------------------------------------
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- metagenomics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file,
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
#> ℹ Final steps .. cleaning & creating back-up
#>
#> ── <metagenomics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 242 features
#> featureData: 7 attributes × 242 features
pcoa_plots <- obj$ordination(metric = "bray",
method = "pcoa",
group_by = "treatment",
weighted = TRUE)
#> 'nperm' >= set of all permutations: complete enumeration.
#> Set of permutations < 'minperm'. Generating entire set.
pcoa_plots
#> $dist
#> S100 S103 S115 S120
#> S100 0.0000000 1.0000000 0.8845188 1.0000000
#> S103 1.0000000 0.0000000 1.0000000 0.9470058
#> S115 0.8845188 1.0000000 0.0000000 1.0000000
#> S120 1.0000000 0.9470058 1.0000000 0.0000000
#>
#> $anova_data
#> pairs Df SumsOfSqs F.Model R2 p.value p.adj
#> 1 tumor vs healthy 1 0.4197984 0.8395968 0.2956747 1 1
#>
#> $pcs
#> PC1 PC2 PC3 groups samples
#> <num> <num> <num> <char> <char>
#> 1: 0.3808548 0.000000e+00 4.422594e-01 tumor 1
#> 2: -0.3808548 -4.735029e-01 -2.256762e-16 tumor 2
#> 3: 0.3808548 -2.787913e-17 -4.422594e-01 healthy 3
#> 4: -0.3808548 4.735029e-01 -2.603956e-16 healthy 4
#>
#> $scree_plot
#> Warning: Removed 7 rows containing missing values or values outside the scale range
#> (`geom_col()`).
 #>
#> $anova_plot
#>
#> $anova_plot
 #>
#> $scores_plot
#> Too few points to calculate an ellipse
#> Too few points to calculate an ellipse
#> Warning: Removed 2 rows containing missing values or values outside the scale range
#> (`geom_path()`).
#>
#> $scores_plot
#> Too few points to calculate an ellipse
#> Too few points to calculate an ellipse
#> Warning: Removed 2 rows containing missing values or values outside the scale range
#> (`geom_path()`).
 #>
## ------------------------------------------------
## Method `omics$foldchange()`
## ------------------------------------------------
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- omics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
obj$scale(method = "clr")
dfe <- obj$foldchange(feature_rank = "Genus",
paired = FALSE,
condition.group = "treatment",
condition_A = c("healthy"),
condition_B = c("tumor"))
#>
#> ── <omics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 64 features
#> featureData: 7 attributes × 64 features
#>
## ------------------------------------------------
## Method `omics$foldchange()`
## ------------------------------------------------
library("ggplot2")
library("OmicFlow")
metadata_file <- system.file("extdata", "metadata.tsv", package = "OmicFlow")
counts_file <- system.file("extdata", "counts.tsv", package = "OmicFlow")
features_file <- system.file("extdata", "features.tsv", package = "OmicFlow")
obj <- omics$new(
metaData = metadata_file,
countData = counts_file,
featureData = features_file
)
#> ✔ metaData template passed the JSON validation.
#> ℹ Checking for duplicated identifiers ..
#> ✔ featureData is loaded.
#> ✔ countData is loaded.
obj$scale(method = "clr")
dfe <- obj$foldchange(feature_rank = "Genus",
paired = FALSE,
condition.group = "treatment",
condition_A = c("healthy"),
condition_B = c("tumor"))
#>
#> ── <omics> object
#> metaData: 9 variables × 4 samples
#> countData: 4 samples × 64 features
#> featureData: 7 attributes × 64 features